X-linked hypophosphatemia (XLH) is a rare bone disease characterized by high levels of FGF23, leading to hypophosphatemia and impaired production of the active form of vitamin D 1,25 dihdroxyvitamin D (1,25D). Patients with XLH have short stature (rickets) and poorly mineralized bones. Their bones have poor quality, in part due to increased resorption of bone surrounding osteocytes and poor connectivity between cells. As adults, they develop an abnormal hardening of the bone-tendon insertion site (enthesis), which causes significant pain and mobility issues. Our laboratory is interested the hormonal and molecular regulation of these complications.
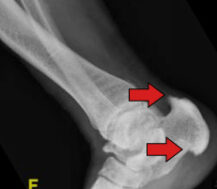
Endocrine regulation of enthesis maturation and pathologic enthesopathy
Patients with XLH develop enthesopathy (mineralized bone-tendon insertion sites). We are interested in learning what signaling pathways modulate the development of enthesopathy and how phosphate and vitamin D regulate these pathways. We also aim to investigate how these signaling pathways and hormones control normal enthesis maturation and they may be involved in the enthesopathies of other musculoskeletal disorders.
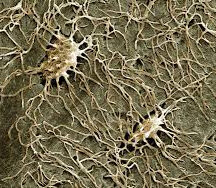
Endocrine regulation of osteocyte lacuno-canalicular remodeling
Osteocytes occupy pits called lacunae and are able to resorb the surrounding mineral matrix. Channels called canaliculi also extend from osteocytes and are necessary for cell-cell communication and transport of small moleculaes. Bones from mice with XLH have significantly abnormal lacunae and canalicular organization. We aim to study how skeletal hormones and signaling pathways regulate the osteocyte mediated remodeling of lacuno-canalicular organization.